what test will confirm a diagnosis of ventricular dilation secondary to dilated cardiomyopathy?
The diagnostic work upwards of genetic and inflammatory dilated cardiomyopathy
An article from the eastward-journal of the ESC Council for Cardiology Do
Vol. 13, N° 19 - 07 Apr 2015
Dilated cardiomyopathy (DCM) is a progressive ventricular wall thinning and dilatation accompanied with gradual functional damage. In daily practice, DCM should non be a concluding diagnosis but rather a ground for farther in-depth investigations. After a presentation of its different forms and treatments, authors offer a simple three-staged diagnostic pathway for DCM patient menstruum.
Background
Although dilated cardiomyopathy (DCM) is far less common than coronary avenue disease and arterial hypertension, DCM is the third cause of centre failure (HF). It typically affects young adults. Numerous conditions and diseases leading to DCM tin be treated effectively nonetheless the diagnostic pathway is difficult and requires comprehensive cognition besides as admission to sophisticated diagnostic methods, such as cardiac magnetic resonance, endomyocardial biopsy and genetic testing. To structure and simplify DCM patient flow, a three-staged diagnostic pathway is proposed.
I - Presentation
Definition - Cardiomyopathy is "a myocardial disorder in which the middle musculus is structurally and functionally abnormal, in the absenteeism of coronary artery disease, hypertension, valvular middle affliction and congenital heart disease sufficient to cause the observed myocardial aberration" (one). Thus, an initial and comprehensive exclusion of those four causes of heart damage and failure which are coronary artery disease, hypertension, valvular center disease and congenital heart affliction should be performed.
Types - Dilated (DCM), hypertrophic (HCM) restrictive (RCM), and arrhythmogenic right ventricular (ARVC) are the four structurally and functionally distinct clinically-orientated classes of cardiomyopathies (2). All the same, types may, as terminate-stage (dilated) HCM, or ARVC with predominantly left ventricular interest often overlap each other or mimic one other.
Cardiomyopathies are further classified into familial and non-familial forms.
Dilated cardiomyopathy - Regardless of the cause of the illness, dilated cardiomyopathy is all-time described every bit a progressive ventricular wall thinning and dilatation accompanied with gradual functional impairment (3). The phenotype of DCM is established by means of imaging studies – echocardiography beingness the most common (4).
Echocardiographic criteria: Echocardiography is used to constitute the type of cardiomyopathy. The cardiomyopathy is considered dilated if the post-obit criteria can be observed 1) left ventricular end-diastolic diameter (LVEDd) > 117% of the age and body surface ii) left ventricular systolic dysfunction (LVSD) defined by left ventricular ejection fraction (LVEF) < 45% and/or three) fractional shortening (FS) < 25%.
In a majority of patients, dilated cardiomyopathy involves the inflammatory process and genetics withal the causes are usually largely unknown.
Epidemiology: The most reliable information on the epidemiology of DCM comes from a study conducted between 1975 and 1984 in Olmsted Canton, Minnesota. Information technology estimated DCM prevalence at 35.5/100,000 inhabitants (five). The annual incidence of DCM is approximately five to 8 per 100,000. DCM is a major cause of sudden death in young adults (6) and is currently the near frequent diagnosis in patients referred for heart transplantation.
Prognosis: Dilated cardiomyopathy is a astringent myocardial disease that results in loftier morbidity and bloodshed, specially in young adults. The course of the affliction is unpredictable, from relatively mild to astringent and rapid, progressing to expiry. Typically, the prognosis is poor with a v-year mortality of 46%.
II – Forms
A) Etiology
The etiology of DCM is heterogeneous. In developed countries CAD and myocardial infarction (MI) are the near common causes of HF, approximating fifty-75% of all HF patients. The terms "non-ischemic cardiomyopathy" have been used to describe dilated cardiomyopathy however they are no longer recommended. Potentially reversible myocardial ischemia must always be sought actively, as effective therapy (medication and revascularisation) tin can favorably alter event. The presence of CAD and MI of form exclude the diagnosis of cardiomyopathy. Despite careful medical history and not-invasive examinations, the causative role of CAD in the development of HF has been shown just later on coronary angiography in upwards to 7 % of patients with initially unexplained DCM (seven).
The phenotype of DCM is common for numerous center conditions. Therefore, the etiology is frequently hard to identify. More often than not, the inflammatory process, inherited forms and idiopathic DCM are equally responsible for more than xc% of cases. Other diagnosable causes of DCM, which always should be considered in the diagnostic procedure, include: remaining types of cardiomyopathies, which may either mimic or progress to DCM, connective tissue diseases, endocrinologic disorders, infiltrative diseases, medications and toxins, tachycardia-induced DCM and lastly miscellaneous causes. Most of these various diseases can exist identified after conscientious medical history, bones laboratory and imaging studies. Even so, some disorders, particularly infiltrative diseases may require more sophisticated examinations, such as laboratory tests, cardiac magnetic resonance (CMR), or the histopathopatological studies (8).
Once these causes of DCM have been excluded, the etiology of DCM can be either genetic (familial) or non-genetic. Further diagnostic steps are based on endomyocardial biopsy and state-of-the-art bioptates cess also equally genetic studies (37). The genetic nature of DCM is increasingly recognised. However, at this phase, only 35% of DCM patients take confirmed causative mutations (9). In the not-genetic causes, persistent myocardial inflammation every bit a consequence of myocarditis is frequent.
B) Familial (genetic)
Idiopathic DCM is considered familial (genetic) when more than i start-degree relative has been diagnosed with DCM. It is estimated that familial DCM can exist diagnosed in xx to over l% of patients with an initial diagnosis of IDC (10). Withal, the true frequency of familial DCM is probably underestimated because some of the initially healthy relatives may develop signs, by and large echocardiographic, and symptoms of the disease during follow-up examinations. Apart from positive three to 4 generation family history and detailed echocardiographic exam of asymptomatic relatives, the phenotype of familial DCM is otherwise duplicate from other forms of DCM. Therefore, confirmation of the causative mutation from detailed genetic analysis and positive family unit history are necessary to establish the diagnosis (nine). The great majority of familial DCM is transmitted in an autosomal potency inheritance pattern; however, other modes of inheritance such as autosomal recessive, X-linked, and mitochondrial have likewise been described (10). So far, numerous mutations in over 30 genes have been identified. The spectrum of genes associated with DCM is broad. Unlike hypertrophic cardiomyopathy which is primarily a disease of sarcomeric proteins, the genetic and molecular basis of DCM is more heterogeneous (11). On the molecular level, a wide diverseness of defected proteins, such as proteins comprising of a nuclear envelope, the cardiac sarcomere, ion channels, transcription factors, and the dystrophin-associated cytoskeletal complex accept been implicated in the pathology of DCM (12, 13). The other unresolved issue is the presence of mutations in typical familial DCM genes in patients who accept neither family history nor echocardiographic abnormalities.
The role of genetic testing in cardiomyopathies has previously been underlined by the American and European Societies. However, the most detailed recommendations on the discipline come up from the 2009 Guidelines of the American Eye Failure Society (fourteen). This certificate provides detailed recommendations for obtaining a family unit history, screening family members, genetic counseling, genetic testing, and handling. Genetic testing for HCM and ARVC is well validated. Withal, genetic evaluation in DCM is also recommended. The other of import issue, addressed past the guidelines, is the genetic overlap betwixt cardiomyopathies. Probably i of the most hitting examples of genetic overlap is DCM and HCM. Whereas in DCM, mutations of sarcomeric proteins cause severe contractile dysfunction, different mutations in the same sarcomeric genes pb to the reverse functional changes that are typically seen in HCM.
C) Not familial (non genetic)
The search for etiology of sporadic cardiomyopathy is even more challenging than familial DCM. Accumulating bear witness, from basic and clinical studies, has paved the way to the concept of inflammation and/or autoimmunisation as the primary cause(due south) of otherwise unexplained cardiomyopathy. Source studies by Felker at al. and from the Myocarditis Treatment Trial take consistently reported myocarditis as the crusade of DCM in ix% and ten% of patients, respectively. Nonetheless, in half of the studied patients with DCM, etiology remained undefined (15). Dilated cardiomyopathy due to inflammation and sub-sequent autoimmunisation is probably more common than was previously thought. Novel imaging software such as CMR with tissue differentiation and techniques of molecular biology used in the assessment of cardiac bioptates, tin can notice inflammatory processes in tissues which would had been previously described as normal (xvi).
D) Inflammatory
Inflammatory dilated cardiomyopathy (DCMI) is a late and serious upshot of the circuitous interplay of the infectious agent, nigh often a virus, and the (auto)-immunologic response, which primarily develops in susceptible individuals (according to a genetic factor) (17). Although the DCMI phenotype is indistinguishable from the typical DCM, the diagnosis of DCMI cannot be made without endomyocardial biopsy (EMB). The state-of-the-art assessment of EMB specimens should encompass non only histo-pathological exam (Dallas criteria) but more importantly immuno-histochemistry and molecular biology techniques to detect DNA or RNA of infectious agent (18- 20). Rarely, myocardial inflammation can be caused past a variety of not-viral infectious organisms including bacteria, fungi, or protozoal agents besides as non-infectious diseases such as systemic diseases, (auto) immune disorders, hypersensitivity reactions, and toxins.
Histological diagnosis of myocarditis (Dallas criteria):
- Active myocarditis – is defined as "an inflammatory infiltrate of the myocardium with necrosis and/or degeneration of adjacent myocytes not typical of the ischemic damage associated with CAD". In most cases infiltrates are mononuclear, but may be neutrophilic, or eosinophilic (in rare cases of eosinophilic myocarditis).
- Borderline myocarditis – is diagnosed when the inflammatory infiltrate is too thin, or myocyte injury is non demonstrated.
Immunohistochemical diagnosis and quantification of inflammatory process (21):
- Abnormal findings – more than 2 CD3+ T-lymphocytes per high-power filed (HPF) (400-fold magnification) or seven per mm2
- Myocarditis – the presence of an inflammatory infiltrate of a minimum of 14 infiltrating leukocytes/mm2
- Agile myocarditis – the presence of > 14 infiltrating leukocytes/mm2 and/or the presence of more than 2 CD3+ T-lymphocytes per HPF, which are adherent to the contour of cardiomyocytes and focally associated with cell necrosis.
The most common cardiotropic viruses that cause myocarditis and DCMI (22):
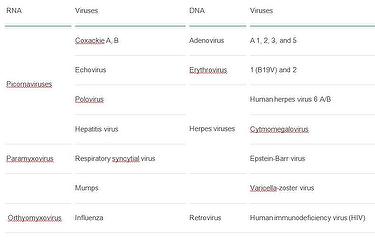
The current theory on the pathogenesis of DCMI states that the affliction is a sequel of an inadequate (too weak or too strong) reaction of host immunologic system directed against microbial agent. More ofttimes DCMI is a consequence of a disproportionately strong reaction of the immunologic system, which escapes from the control mechanisms despite numerous "switching off" signals. In the prolonged low-level inflammation, cardiomyocytes are phagocytised, surrounded by various pathologic auto-antibodies, and pushed towards apoptosis. There is imbalance of pro-inflammatory cytokines, such every bit tumor necrosis factor (TNFα) or interleukins – IL-6 and IL-17 over anti-inflammatory cytokines tumor growth factor (TGFβ) or interferon (IFNγ) (23). Equally a result, the number of cardiomyocytes is greatly reduced.
III - 3-staged diagnostic process
To construction and simplify the DCM patient menstruation, a three-staged diagnostic algorithm has been proposed and is presented below.
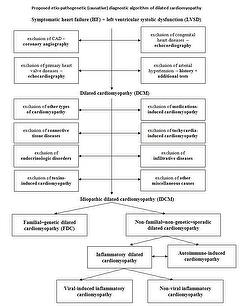
Phase I – Screening
- Exclusion of coronary artery illness (CAD)
Significant CAD is most ordinarily divers as > l% stenosis of at to the lowest degree one main coronary artery. - Exclusion of primary valvular heart disease or congenital heart illness
This should be performed by history taking, medical records, physical examinations and peculiarly past transthoracic echocardiography and if necessary by - Exclusion of arterial hypertension
Exclusion of hypertension is probably the almost challenging in this initial diagnostic process, particularly in individuals more advanced in historic period. Tools to verify the possible role of hypertension in the pathogenesis of DCM should be a careful patient history, including the number of drugs and the dosage of antihypertensive medications, previous hospital records, every bit well as electrocardiographic and echocardiographic signs of LV hypertrophy.
Stage Ii – Identification of "diagnosable" causes
- Other forms of cardiomyopathies that may mimic and/or lead to DCM include: finish-stage HCM, RCM, peripartum cardiomyopathy, stress-induced cardiomyopathy (Tako-tsubo syndrome), left ventricular non-compaction (24- 26). The presence of singled-out features of the same types of cardiomyopathies is verified by comprehensive echocardiography and CMR.
- Connective tissue diseases include systemic lupus erythematosous, scleroderma, giant cell arthritis, etc. Exclusion procedure depends mostly on typical features of connective tissue disorders as well as the presence and titer of antinuclear and other disease-specific antibodies.
- Endocrinologic disorders that may rarely lead to the phenotype of DCM include thyroid hormone excess or deficiency, pheochromocytoma, Cushing's disease.
- Toxins-induced cardiomyopathy – the most common toxin is ethanol following past illegal drug abuse of cocaine and amphetamines. Other rare toxins include cobalt, atomic number 82, mercury. Detailed patient's history is the about important component.
- Medications-induced cardiomyopathy – the most pathogenic are chemotherapeutic agents such as anthracyclines, cyclophosphamide, and trastuzumab. Additionally, antiretroviral drugs (against HIV infection) such every bit zidovudine, didanosine, and zalcitabine are proven drugs implicated in DCM. Less evidence exists for other medications phenothiazines, chloroquine, and clozapine.
- Tachycardia-induced cardiomyopathy – persistent tachycardia, particularly uncontrolled atrial fibrillation, atrio-ventricular nodal reentry, and preexcitation syndromes with ventricular rates of 130 to 200 beats per minute may lead to DCM.
- Infiltrative disease – include amyloidosis, sarcoidosis, and hemochromatosis. Amyloidosis produces typical signs on ECG (depression voltage in the limb and chest leads and conduction abnormalities), echocardiography (prominent diastolic dysfunction, symmetric LV wall thickening, and a granular, "sparkling" appearance of the myocardium), and amyloid deposits on myocardial bioptates. Diagnosis of sarcoidosis depends on the multi-arrangement features of sarcoidosis and the testify of not-caseating granulomas isolated either from the myocardium or other affected tissue.
- Hemochromatosis should be suspected by the presence of an elevated serum transferrin saturation and confirmed after typical findings of very depression bespeak intensity on CMR and/or detected atomic number 26 deposits on cardiac samples.
- Miscellaneous causes of cardiomyopathy include neuromuscular diseases (Duchenne's muscular dystrophy, myotonic dystrophy, Friedreich's ataxia), neoplastic heart disease (primary heart tumors, metastases to center), celiac disease, extensive breast radiation, nutritional deficiencies (thiamine, selenium, and 50-carnitine), obstructive sleep apnea (27). Those causes are very rare, notwithstanding when suspected, appropriate diagnostic pathways should be followed.
At the end of Stage II, DCM volition exist classified as idiopathic dilated cardiomyopathy as no other detectable causes of DCM have been identified.
Stage III – Establishing the etiology of idiopathic dilated cardiomyopathy
Appropriate examinations such as echocardiographic studies of family members, genetic studies, and histopathological/immunochemistry/molecular biology studies of cardiac samples should be performed to differentiate between 2 master causes of idiopathic DCM such as genetic and inflammation/autoimmunisation.
Conclusions
A simplified three-staged diagnostic pathway has been proposed. After exclusion of the relatively rare causes of DCM, such as endocrinologic or autoimmune disorders, there are two wide groups of remaining weather – genetic DCM and inflammatory DCM which are usually a distant sequel of myocarditis. In specialised cardiomyopathy centers, sophisticated in-depth diagnosis of genetic and inflammatory causes of DCM are slowly condign a reality. Unfortunately, at present evidence-based and cost-effective causative treatments of genetic or inflammatory DCM remain difficult. This is yet a very dynamic field and with possible therapeutic improvements.
The point is not to advocate complicated and expensive tests for every DCM patient, yet, this is a very dynamic field and we should exist aware of the growing diagnostic potential and possibly novel effective treatment that may be but around the corner for what were once "no hope" patients.
The content of this commodity reflects the personal opinion of the author/south and is not necessarily the official position of the European Order of Cardiology.
References
- Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ, Monserrat 50, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Eur Heart J. 2008 Jan;29(two):270-6
- Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and last diagnosis. A position statement from the ESC Working Grouping on Myocardial and Pericardial Diseases. Rapezzi C, Arbustini Eastward, Caforio AL, Charron P, Gimeno-Blanes J, Heliö T, Linhart A, Mogensen J, Pinto Y, Ristic A, Seggewiss H, Sinagra G, Tavazzi 50, Elliott PM. Eur Heart J. 2013 May;34(19):1448-58. doi: 10.1093/eurheartj/ehs397.
- ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Center Failure Association (HFA) of the ESC. McMurray JJ1, Adamopoulos Southward, Anker SD, Auricchio A, Böhm M, Dickstein Chiliad, Falk V, Filippatos One thousand, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A; Eur Heart J. 2012 Jul;33(14):1787-847. doi: ten.1093/eurheartj/ehs104.
- Recommendations for chamber quantification: a report from the American Society of Echocardiography'southward Guidelines and Standards Commission and the Bedroom Quantification Writing Group, developed in conjunction with the European Clan of Echocardiography, a branch of the European Society of Cardiology. Lang RM1, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ; Chamber Quantification Writing Group; American Society of Echocardiography'southward Guidelines and Standards Committee; European Association of Echocardiography. J Am Soc Echocardiogr. 2005 December;eighteen(12):1440-63.
- Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based report in Olmsted Canton, Minnesota, 1975-1984. Codd MB1, Sugrue DD, Gersh BJ, Melton LJ tertiary. Circulation. 1989 Sep;80(3):564-72.
- Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: changing bloodshed over the last 30 years. Merlo M1, Pivetta A, Pinamonti B, Stolfo D, Zecchin Chiliad, Barbati G, Di Lenarda A, Sinagra Yard. Eur J Heart Neglect. 2014 Mar;16(3):317-24. doi: 10.1002/ejhf.xvi.
- Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. Felker GM1, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. N Engl J Med. 2000 Apr xiii;342(15):1077-84.
- Inherited cardiomyopathies. Watkins H1, Ashrafian H, Redwood C. N Engl J Med. 2011 Apr 28;364(17):1643-56. doi: 10.1056/NEJMra0902923.
- Familial dilated cardiomyopathy: prove for genetic and phenotypic heterogeneity. Heart Musculus Disease Study Grouping. Mestroni L1, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, Vatta Thou, Pinamonti B, Muntoni F, Caforio AL, McKenna WJ, Falaschi A, Giacca M, Camerini. J Am Coll Cardiol. 1999 Jul;34(1):181-xc.
- Recent progress in the genetics of cardiomyopathy and its role in the clinical evaluation of patients with cardiomyopathy. Ghosh N1, Haddad H. Curr Opin Cardiol. 2011 Mar;26(2):155-64. doi: 10.1097/HCO.0b013e3283439797.
- The function of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Møller DV1, Andersen PS, Hedley P, Ersbøll MK, Bundgaard H, Moolman-Smook J, Christiansen One thousand, Køber L. Eur J Hum Genet. 2009 October;17(10):1241-9. doi: 10.1038/ejhg.2009.34.
- Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Centre Failure. Waldmüller S1, Erdmann J, Binner P, Gelbrich G, Pankuweit S, Geier C, Timmermann B, Haremza J, Perrot A, Scheer S, Wachter R, Schulze-Waltrup Northward, Dermintzoglou A, Schönberger J, Zeh W, Jurmann B, Brodherr T, Börgel J, Farr M, Milting H, Blankenfeldt Due west, Reinhardt R, Özcelik C, Osterziel KJ, Loeffler Yard, Maisch B, Regitz-Zagrosek Five, Schunkert H, Scheffold T; German Competence Network Eye Failure. Eur J Heart Fail. 2011 Nov;thirteen(11):1185-92. doi: 10.1093/eurjhf/hfr074.
- Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Parks SB1, Kushner JD, Nauman D, Burgess D, Ludwigsen Due south, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Am Center J. 2008 Jul;156(1):161-nine. doi: ten.1016/j.ahj.2008.01.026.
- Genetic evaluation of cardiomyopathy--a Heart Failure Gild of America practise guideline. Hershberger RE1, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA; Centre Failure Gild of America. J Carte du jour Fail. 2009 Mar;15(2):83-97. doi: 10.1016/j.cardfail.2009.01.006.
- A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Handling Trial Investigators. Mason JW1, O'Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, Moon TE. North Engl J Med. 1995 Aug iii;333(five):269-75.
- Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. Friedrich MG1, Sechtem U, Schulz-Menger J, Holmvang Yard, Alakija P, Cooper LT, White JA, Abdel-Aty H, Gutberlet M, Prasad S, Aletras A, Laissy JP, Paterson I, Filipchuk NG, Kumar A, Pauschinger One thousand, Liu P; International Consensus Grouping on Cardiovascular Magnetic Resonance in Myocarditis. J Am Coll Cardiol. 2009 Apr 28;53(17):1475-87. doi: x.1016/j.jacc.2009.02.007.
- Current country of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu Yard, Heliö T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi Fifty, Thiene G, Yilmaz A, Charron P, Elliott PM; European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013 Sep;34(33):2636-48, 2648a-2648d. doi: ten.1093/eurheartj/eht210.
- The office of endomyocardial biopsy in the direction of cardiovascular illness: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology Endorsed by the Heart Failure Society of America and the Heart Failure Clan of the European Society of Cardiology. Cooper LT1, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R. Eur Heart J. 2007 Dec;28(24):3076-93.
- Myocarditis: the Dallas criteria. Aretz HT. Hum Pathol. 1987 Jun;18(6):619-24.
- Diagnosis of myocarditis: death of Dallas criteria. Baughman KL. Circulation. 2006 January 31;113(iv):593-five.
- World Middle Federation consensus conference's definition on inflammatory cardiomyopathy (myocarditis): report from 2 skilful committees on histology and viral cardiomyopathy. Maisch B, Bultman B, Factor South. Heartbeat 1999; 4; 3-four.
- The management of myocarditis. Schultheiss HP1, Kühl U, Cooper LT. Eur Middle J. 2011 Nov;32(21):2616-25. doi: 10.1093/eurheartj/ehr165.
- Presentation, patterns of myocardial harm, and clinical course of viral myocarditis. Mahrholdt H1, Wagner A, Deluigi CC, Kispert E, Hager South, Meinhardt One thousand, Vogelsberg H, Fritz P, Dippon J, Bock CT, Klingel One thousand, Kandolf R, Sechtem U. Circulation. 2006 Oct 10;114(fifteen):1581-xc.
- American College of Cardiology/European Gild of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Strength on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Maron BJ1, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH 3rd, Spirito P, X Cate FJ, Wigle ED; American Higher of Cardiology Foundation Task Force on Clinical Practiced Consensus Documents; European Order of Cardiology Committee for Do Guidelines.Eur Heart J. 2003 Nov;24(21):1965-91.
- Current land of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Middle Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Sliwa Yard, Hilfiker-Kleiner D, Petrie MC, Mebazaa A, Pieske B, Buchmann Due east, Regitz-Zagrosek V, Schaufelberger One thousand, Tavazzi L, van Veldhuisen DJ, Watkins H, Shah AJ, Seferovic PM, Elkayam U, Pankuweit S, Papp Z, Mouquet F, McMurray JJ; Heart Failure Association of the European Society of Cardiology Working Group on Peripartum Cardiomyopathy. Eur J Heart Fail. 2010 Aug;12(8):767-78. doi: 10.1093/eurjhf/hfq120.
- Left ventricular not-compaction revisited: a distinct phenotype with genetic heterogeneity? Oechslin E, Jenni R. Eur Middle J. 2011 Jun;32(12):1446-56. doi: 10.1093/eurheartj/ehq508.
- Cardiomyopathy of Friedreich ataxia. Weidemann F, Störk S, Liu D, Hu K, Herrmann S, Ertl G, Niemann M. J Neurochem. 2013 Aug;126 Suppl 1:88-93. doi: 10.1111/jnc.12217.
Notes to editor
Dr Pawel Rubis
Section of Cardiac and Vascular Affliction, John Paul II Infirmary, Institute of Cardiology, Krakow, Poland
Author's disclosures: None declared.
The content of this article reflects the personal opinion of the writer/s and is not necessarily the official position of the European Society of Cardiology.
Source: https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-13/The-diagnostic-work-up-of-genetic-and-inflammatory-dilated-cardiomyopathy
0 Response to "what test will confirm a diagnosis of ventricular dilation secondary to dilated cardiomyopathy?"
Post a Comment